INTUSUSCEPCIÓN INTESTINAL MÁS GRANDE REPORTADA
EN PACIENTE CON SÍNDROME DE PEUTZ-JEGHERS. INFORME DE CASO
Miguel Vassallo P.1, Alexis Oliveros C.2, Sailú Bravo3, Carlos Acero4, Bárbara Durán5, Daniela Vieira5
1. Cirujano General. Jefe de servicio Cirugía II Hospital Universitario de Caracas. Caracas-Venezuela
2. Cirujano General. Hospital Universitario de Caracas. Caracas- Venezuela
3. Cirujano General. Profesor instructor
Cirugía II Escuela de medicina Luis Razetti Hospital
Universitario de Caracas UCV.
4. Residente de cuarto año
Cirugía General. Cirugía II Hospital Universitario de
Caracas. Caracas- Venezuela
5. Estudiante de 5to año de Medicina,
Escuela ¨Luis Razetti¨. Universidad Central de Venezuela.
Caracas- Venezuela. Correo-e: barbaramaria0411@gmail.com
RESUMEN
Introducción: La intususcepción intestinal se considera
una causa rara de obstrucción intestinal en adultos, sus efectos
son graves y a menudo requieren intervención quirúrgica,
su diagnóstico es un reto dada sus manifestaciones
clínicas inespecíficas. El síndrome de
Peutz-Jeghers es una enfermedad genética poco frecuente,
caracterizada por la presencia de pólipos hamartomatosos
intestinales y pigmentaciones mucocutáneas. Durante su
crecimiento dichos pólipos pueden llegar a complicarse y causar
intususcepción, obstrucción y hemorragias intestinales.
Caso clínico: femenina de 18 años con diagnóstico
de SPJ, quien refiere inicio de enfermedad actual 13 días
previos a su ingreso, caracterizado por dolor tipo cólico de
moderada intensidad en epigastrio, evacuaciones líquidas, en
número de 5 al día, sin moco ni sangre, náuseas y
vómitos de contenido alimentario en múltiples
oportunidades. Se realizó US abdominal evidenciando imagen
redondeada en epigastrio hipoecoica de 78.4mm x 49.3mm. Fue llevada a
mesa operatoria bajo el diagnóstico de Abdomen agudo Obstructivo
secundario a: Intususcepción. Los hallazgos quirúrgicos
reportaron un Intusuceptum yeyunal de 60cm con cambios
isquémicos irreversibles, por lo que se decide realizar
resección y anastomosis, evolucionando de forma satisfactoria
Conclusión: Evaluando la literatura disponible la
intususcepción intestinal más larga hasta ahora reportada
es de 50 cm por lo tanto nuestro trabajo pretende aportar nueva
información y memoria académica sobre esta entidad
clínica.
Palabras clave: Caso clínico, Síndrome de Peutz-Jeghers,
Intususcepción, Obstrucción Intestinal, Poliposis
Intestinal
Larger intestinal intussusception reported in a patient with Peutz-Jeghers syndrome. Case report
ABSTRACT
Introduction: Intestinal intussusception is considered a rare cause of
intestinal obstruction in adults, its effects are serious and often
require surgical intervention, its diagnosis is a challenge given its
non-specific clinical manifestations. Peutz-Jeghers syndrome is a rare
genetic disease, characterized by the presence of intestinal
hamartomatous polyps and mucocutaneous pigmentations. During their
growth, these polyps can become complicated and cause intussusception,
obstruction and intestinal bleeding. Clinical case: of an 18-year-old
female with a diagnosis of PJS, who reported the onset of current
illness 13 days prior to her admission, characterized by colicky pain
of moderate intensity in the epigastrium, liquid stools, 5 per day,
without mucus. nor blood, nausea and vomiting of food content on
multiple occasions. An abdominal US was performed showing a rounded
image in the hypoechoic epigastrium measuring 78.4mm x 49.3mm. She was
taken to the operating table under the diagnosis of Acute Obstructive
Abdomen secondary to: Intussusception. The surgical findings reported a
jejunal intussusception of 60cm with irreversible ischemic changes, so
it was decided to perform resection and anastomosis, progressing
satisfactorily Conclusion: Evaluating the available literature, the
longest intestinal intussusception reported so far is 50cm, therefore
our work aims to provide new information and academic memory about this
clinical entity.
Key words: Clinical case, Peutz-Jeghers Syndrome, Intussusception, Intestinal Obstruction, Intestinal Polyposis
INTRODUCCIÓN
El Síndrome de Peutz-Jeghers (SPJ) es una enfermedad hereditaria
genética autosómica dominante, poco frecuente,
caracterizada por máculas hiperpigmentadas melánicas
pardas o azul oscuras típicamente en los labios y la mucosa
bucal, aunque también se pueden presentar en la cara, manos,
pies, paladar, región perianal y vagina, asociado con
Pólipos Hamartomatosos (PH) del tracto gastrointestinal.(1) El
50% de los pacientes afectados con SPJ presentarán
manifestaciones clínicas antes de los 20 años.(2) Se ha
demostrado que las mutaciones en el gen supresor de tumores
Serina/Treonina quinasa 11 (STK11) en el cromosoma 19p13 causa este
síndrome.(2,3) .La intususcepción Intestinal (II) es la
afección en la que parte del intestino se desliza con su pliegue
mesentérico en la luz del intestino distal adyacente, alterando
la peristalsis, obstruyendo el paso libre del contenido intestinal,
comprometiendo el flujo vascular mesentérico de este segmento y
finalmente resultando en OI.(4)
En la bibliografía actual la intususcepción más
larga antes reportada posee una medida de 50 cm.(4) Se realizó
una búsqueda exhaustiva utilizando palabras claves como,
“invaginación”, “intususcepción”,
“SPJ”, en los buscadores Pubmed, Cochrane, Medline, Scielo,
ChatGTP, Tridatabase, no encontrándose ningún otro caso
que superara esas medidas. A continuación, presentamos el caso
de un paciente con hallazgo intraoperatorio de una II de 60 cm.
Presentación del caso
Información del Paciente
Femenina de 18 años, con diagnóstico de SPJ a los 5
años, antecedentes quirúrgicos de polipectomía
endoscópica: Gástrica, duodenal, cecal y de colon
transverso, sin complicaciones, reportando ciego: Pólipo de
Peutz Jeghers con displasia de bajo grado. Polipectomía por
Histeroscopia realizada sin complicaciones, sin biopsia. Refiere IEA 13
días previos a su ingreso caracterizado por dolor abdominal tipo
cólico de aparición insidiosa, en epigastrio de moderada
intensidad, irradiado a mesogastrio, que atenúa parcialmente con
ingesta de AINES, concomitantes evacuaciones líquidas, en
número de 5 al día, sin moco ni sangre, náuseas y
vómitos de contenido alimentario en múltiples
oportunidades. Por persistencia de sintomatología acude al HUC,
donde es evaluada e ingresada.
Al examen físico, signos vitales: Frecuencia cardíaca 150
lpm, Frecuencia respiratoria 30 rpm, Presión arterial 100/65
mmHg, Sat O2 98%. Fascies álgida, posición
antálgica en decúbito lateral derecho. Palidez
cutáneo mucosa, sequedad de piel y mucosas. Abdomen distendido,
Ruidos hidroaéreos ausentes, poco depresible, doloroso a la
palpación superficial y profunda con signo de Gueneau de Mussy
positivo.
Laboratorio reporta anemia severa (Hemoglobina 7,4 gr/dL) y leucocitosis a predominio de segmentados.
Ultrasonografía abdominal: Imagen redondeada hipoecoica en
epigastrio de bordes bien definidos con zona central ecogénica
de 78.4mm x 49,3 mm.
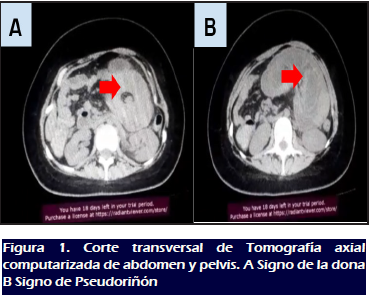
Tomografía abdominal: Múltiples lesiones nodulares
polipoideas intraluminales con densidad de tejidos blandos, los cuales
comprometen el estómago, yeyuno e íleon, evidenciando
signo de la “dona” (Figura 1A),
“pseudoriñón” (Figura 1B),
características de la II.
SEGUIMIENTO Y RESULTADOS
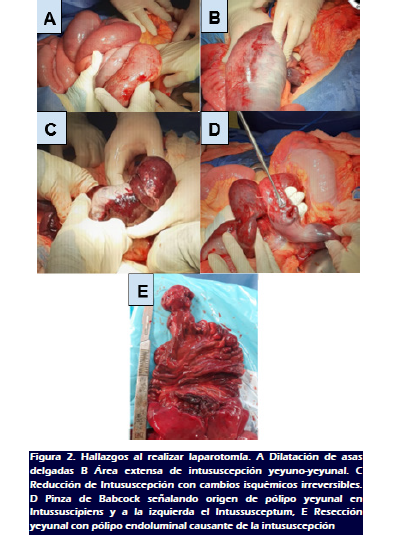
Se realizó laparotomía exploradora a cargo de un R3 junto
a un cirujano con experiencia encontrando: 600 cc de líquido
inflamatorio libre en cavidad, vólvulo de 100 cm de asa delgada
dilatada con cambios isquémicos irreversibles (Figura 2A), a
partir de ángulo duodenoyeyunal, Intususceptum yeyunal de 60cm
necrótico (Figura 2B), Pólipo endoluminal en yeyuno a 4
cm y 70 cm de asa fija (Figura 2C), Pólipo endoluminal no
obstructivo en íleon a 5 cm de válvula ileocecal. Se
decidió realizar desvolvulación intestinal,
reducción de intususcepción (Figura 2 C,D),
resección segmentaria de 110 cm de yeyuno a partir de asa fija,
y entero-entero anastomosis término terminal. (Figura 2E). No
hubo complicaciones intra ni postoperatorias.
La biopsia de la pieza quirúrgica reportó: Pólipo
hiperplásico asentado sobre mucosa yeyunal con
inflamación crónica moderada a severa, activa,
irregularmente distribuida sobre proliferación de músculo
liso. Necrosis yeyunal extensa.
Por evolución satisfactoria, es egresada al séptimo día postoperatorio.
DISCUSIÓN
El SPJ es una afección poco frecuente que se presenta sin
distinción de género ni predominio racial, caracterizado
por PH en el tracto gastrointestinal, con una prevalencia: intestino
delgado de 64%, colon 53.2%, estómago 48%, y recto 32%. Aunque
pueden encontrarse pólipos extraintestinales.(5)
Estas lesiones tienden a desarrollarse durante la pubertad y se
manifiestan sintomáticamente entre los 10 y 30 años, con
síntomas que incluyen dolor abdominal, OI, hemorragias
digestivas, anemia e II. (1,3,5)
La II se define como el prolapso o telescoping de un segmento
intestinal en otro segmento adyacente. Aproximadamente la mitad de los
pacientes con SPJ experimentaran una intususcepción a lo largo
de su vida. En adultos, la II tiene una incidencia baja, representando
el 1-5% de los casos de OI mecánica. En el SPJ, los
pólipos desempeñan un papel crucial al actuar como puntos
de anclaje que, al ser desplazados por el peristaltismo intestinal,
provocan su inserción en el segmento adyacente del intestino.
(5) Esta entidad puede observarse en diferentes modalidades de imagen,
por ecografía puede identificarse el ‘signo de la
dona’ al obtener imágenes del eje corto del segmento
comprometido o puede verse como un pseudoriñón al obtener
imágenes longitudinales. En tomografía se revela como una
masa de tejidos blandos compleja cuando se observa centralmente el asa
intususceptum y periféricamente el asa intususcipiens, pueden
verse porciones de baja densidad que corresponden a grasa
mesentérica asociadas a vasos mesentéricos
acompañando el intususceptum. Siendo esta última el gold
estándar para el diagnóstico con sensibilidad y
especificidad del 85 al 100%. (1,3,5,6)
Durante la exploración quirúrgica, se identificó
un intususceptum yeyunal de 60 cm necrótico. En la literatura
actual, la intususcepción más extensa documentada es de
50 cm, mientras que la encontrada en nuestra paciente alcanzó
los 60 cm, marcando un caso excepcional en términos de longitud.
Dado la baja prevalencia de esta patologia, no existe un protocolo de
manejo estandarizado. La elección del procedimiento se basa en
consideraciones como la ubicación, tamaño, causa y
viabilidad del intestino. Según las directrices del Grupo
Europeo de Tumores Hereditarios (EHTG), se recomienda realizar la
reducción quirúrgica de la intususcepción de
manera inmediata para prevenir la necrosis y evitar grandes resecciones
intestinales. En general, la laparotomía es la técnica
principal, junto con la resección del pólipo causante del
intussusceptum mediante enterotomía, y la evaluación
detallada de todo el intestino, con la resección de todos los
pólipos mayores de 15 mm mediante enterotomía o
enteroscopia intraoperatoria. (6)
La vigilancia periódica y la extirpación de
pólipos reducen la probabilidad de complicaciones, especialmente
la Il, además de la necesidad de intervenciones
quirúrgicas y resecciones intestinales.
CONCLUSIÓN
En conclusión, el SPJ es una patología rara asociada a
diversas complicaciones, siendo la II una de las más destacadas.
Esta puede presentarse de manera inespecífica, dificultando el
diagnóstico y aumentando el riesgo de isquemia intestinal,
incrementando la tasa de mortalidad. Es de vital importancia que el
cirujano mantenga un alto índice de sospecha para realizar
diagnósticos certeros y oportunos, proporcionando así la
mejor atención y manejo al paciente.
Aprobación ética: Se obtuvo el consentimiento informado del paciente incluido en el estudio.
Conflicto de Intereses
Los autores declaran no tener conflicto de interés alguno sobre el presente estudio.
REFERENCIAS
1. Ospina Nieto J, , Pío Quintero Á.
Síndrome de Peutz-Jeghers. Presentación de casos y
revisión de la literatura. Revista Colombiana de
Gastroenterología.2009;24(2):188-199.
https://www.redalyc.org/articulo.oa?id=337731592013
2. Achatz, M. I., Porter, C. C., Brugières, L., Druker,
H., Frebourg, T., Foulkes, W. D., … Plon, S. E. (2017). Cancer
screening recommendations and clinical management of inherited
gastrointestinal cancer syndromes in childhood. Clinical Cancer
Research: An Official Journal of the American Association for Cancer
Research, 23(13), e107–e114.
https://doi:10.1158/1078-0432.ccr-17-0790
3. Orphanet: Síndrome de Peutz-Jeghers. 2020. https://www.orpha.net/es/disease/detail/2869
4. Kalliakmanis V, Perysinakis I, Koutsouvas K, Karras P,
Margaris E, Angelakis C. Massive intussusception caused by a solitary
Peutz–Jeghers type hamartomatous polyp. Ann R Coll Surg Engl
2018;100:e91–3. https://doi.org/10.1308/rcsann.2018.0019.
5. Síndrome de Peutz-Jeghers. (2010).
Gastroenterología y hepatología continuada, 9(2),
55–59. https://doi:10.1016/s1578-1550(10)70012-7
6. Wagner, A., Aretz, S., Auranen, A., Bruno, M. J., Cavestro,
G. M., Crosbie, E.J., Möslein, G. (2021). The management of
Peutz–Jeghers syndrome: European hereditary tumour group (EHTG)
guideline. Journal of Clinical Medicine, 10(3), 473.
https://doi:10.3390/jcm10030473.